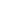2016-08-12 儿研所
近日,我院儿研所基因检测平台成功诊断假肥大型肌营养不良(DMD)患者2例。在第一例DMD基因检测中,先证者,男,6岁半,于今年因站立和行走困难,动作笨拙,易跌倒,走路摇摇晃晃,登楼梯或由坐、卧位起立困难,臀中肌无力导致行走时呈特殊的鸭步等症状就诊于我院儿保科,经常规生化检测发现CK值高达12000U/L ,被怀疑患有DMD。我院儿研所基因检测平台经采用MLPA的检测方法对患儿及其母亲的DMD基因的大片段缺失和重复突变进行了检测和分析。检测结果显示患儿的假肥大性肌营养不良致病基因DMD外显子46-48发生纯合缺失突变,其母亲DMD基因外显子46-48发生了杂合缺失突变。并且该突变方式的致病性已有文献报道,符合假肥大性肌营养不良的基因突变特征。同时,我院儿研所杨颖博士对于生育下一代的患病风险及如何避免出生缺陷给予了患儿家长详细的介绍和解释。
在第二例DMD基因检测中,母亲已于当地医院检测到她的DMD基因外显子52杂合缺失,其弟弟早年因DMD已去世。考虑到DMD属X连锁隐性遗传,她非常忧心是否会遗传给自己的儿子,情绪非常焦虑。其子3岁,无临床症状,CK值在正常范围内。为了检测其子是否遗传到DMD外显子52缺失,我院儿研所基因检测平台采用MLPA的检测方法对患儿的DMD基因的大片段缺失和重复突变进行了检测和分析。检测结果显示其子DMD基因并未缺失,该检测结果也大大缓解了该母亲的忧心与担忧。
假肥大性肌营养不良(DMD),也称 Duchenne型肌营养不良症(DMD),是最常见的一类进行性肌营养不良症。患病率为 3.3/10万,占出生男婴的20-30/10万,为X-连锁隐性遗传。主要是男孩发病,女性为致病基因的携带者。通常5岁左右发病。肌萎缩是进行性的,这是本病的特点,预后差。本病的发生是由于编码dystrophin的DMD基因的突变所引起。
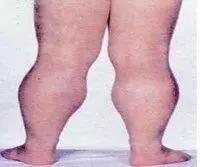
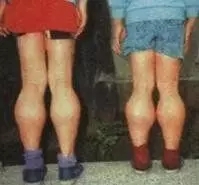
患儿出生时的活动如抬头、坐姿等均正常,自 1 岁以后开始逐渐出现站立和行走困难,首先影响骨盆带肌肉,以后累及肩胛带肌肉。患儿动作笨拙,易跌倒,走路摇摇晃晃,登楼梯或由坐、卧位起立困难。随着病变的进展,臀中肌无力导致行走时呈特殊的鸭步,患儿从仰卧位起立时需先翻转为俯卧,再以双手支持地面和下肢缓慢地站立。约 1/4 的患儿有智力低下,血中磷酸肌酸激酶( CRK )浓度显著升高,可达正常值的 1万倍。病变呈进行性加重,常到 10 岁时已不能行走,大多数患儿最终卧床不起,并发痉挛、褥疮、肺炎而在 20 岁前死亡。因此,在已生育过 DMD患者的家庭中,如果能应用基因诊断方法,对所怀男胎排除DMD基因的突变,仍可生育正常男胎。
自今年起,我院儿研所基因检测平台逐渐完善,目前已经成功诊断多例单基因遗传病,近期新开展DMD以及脊髓性肌萎缩(spinalmuscularatrophy,简称SMA)基因检测,将给予临床诊断提供有力的科学依据,为患儿家长提供更多的有关于优生优育方面的信息。
信息来源: 西安市儿童医院